Research
The discovery that RNA can act as a catalyst continues to have far reaching impacts on evolution, synthetic biology, and diagnostics. Ongoing revolutions in transcriptomics, metabolomics, structural biology, and computation are fueling a rapid expansion in our understanding of the myriad of RNA-protein interactions that are essential for gene expression. Nonetheless, this rapid expansion has exposed two critical gaps in our knowledge that restrict our ability to learn and apply basic principles of RNA catalysis and specificity to understand biology and limit applications of RNA in therapeutics and drug discovery.
First, we lack sufficient atomic-level experimental information to accurately model the mechanisms and transition states of RNA reactions, which presents a critical barrier to defining principles of biological phosphoryl transfer for enzyme engineering and inhibitor design. Second, the broad specificity of most RNA binding proteins and RNases cannot be described in simple binary terms (i.e., specific versus non-specific), requiring new paradigms for understanding their function. Our research addresses these critical gaps in our knowledge by applying innovative experimental approaches, combined with traditional computational, biochemical, and structural tools. Our long-term vision is to make a fundamental leap forward in our understanding of biological phosphoryl transfer, drive the field toward quantitative and comprehensive concepts of RNA specificity, and realize novel opportunities in drug discovery.
Mechanistic Enzymology of Ribozyme Catalysis
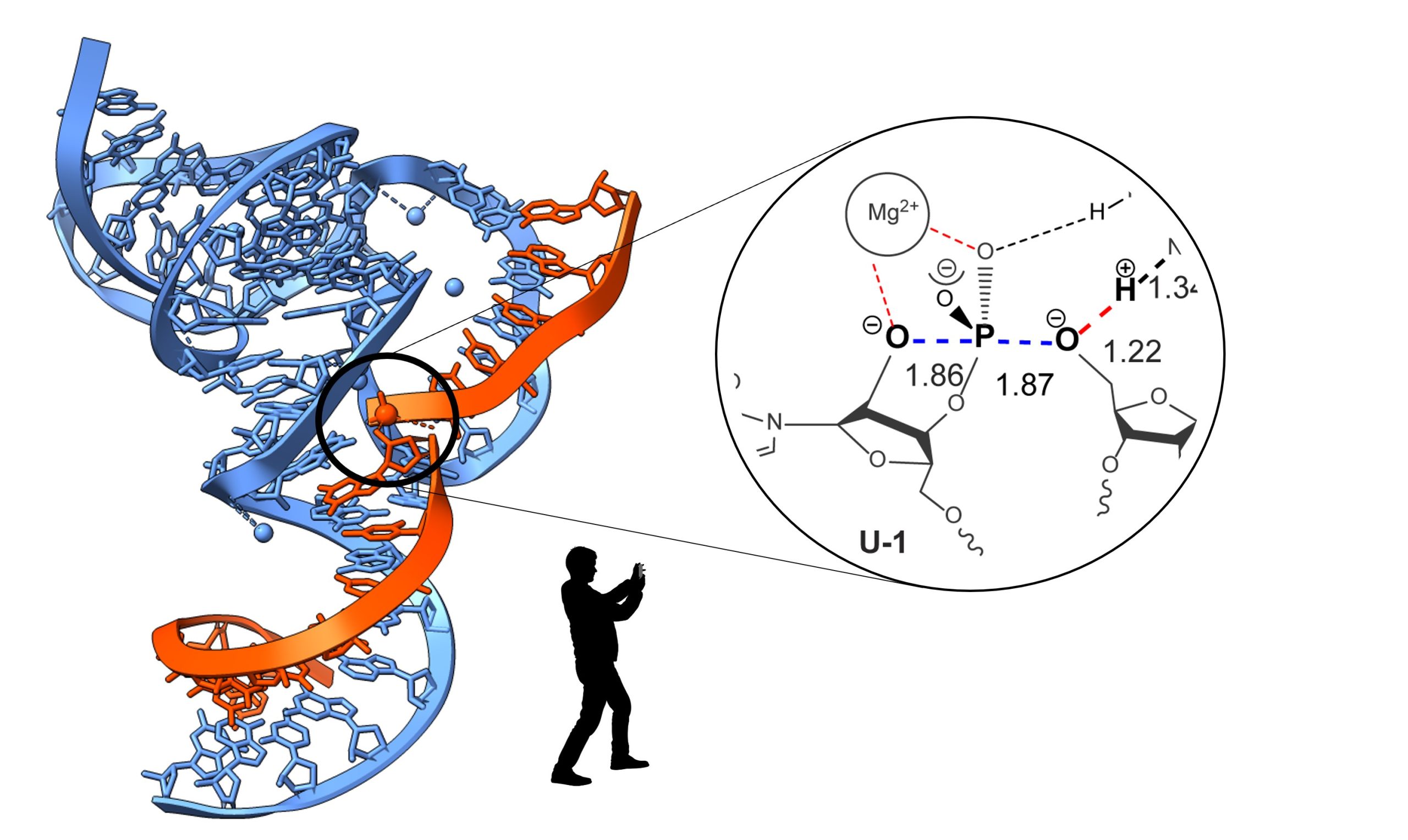
RNA strand cleavage by 2’-O-transphosphorylation is a ubiquitous reaction in biology that is catalyzed by both protein ribonucleases and ribozymes. Thus, comparative studies of RNA cleavage by RNA and protein provide the opportunity to define general principles of biological catalysis18,19 that can guide enzyme engineering and inhibitor design. Structural and biochemical studies have demonstrated the essential roles of active site acids, bases, and metal ions in transition state stabilization. New and powerful computational methods have yielded highly detailed atomistic models of the mechanistic pathways of RNA catalysis. However, a critical barrier to evaluating these models is lack of experimental information on transition state structure.
The best, and arguably the only, experimental approach for analyzing enzymatic TSs is analyses of kinetic isotope effects (KIEs). An important step forward was our development in collaboration with the labs of Dr. Darrin York and Dr. Joseph Piccirilli of an approach that integrates KIEs with multi-scale simulations to determine the mechanisms and TSs of RNA reactions. Recently, we applied our integrated experimental and computational approach to demonstrate that RNases and ribozymes can direct phosphoryl transfer reactions along surprisingly different mechanistic pathways with distinct TS structures. Now, we aim to discover how active site interactions enforce alternative reaction paths for phosphoryl transfer, extend our adaptable approach for KIE measurements to additional biomedically important phosphoryl transfer enzymes, and ultimately pursue new opportunities in TS-based inhibitor design.
Substrate Specificity and Inhibition of Bacterial RNase P
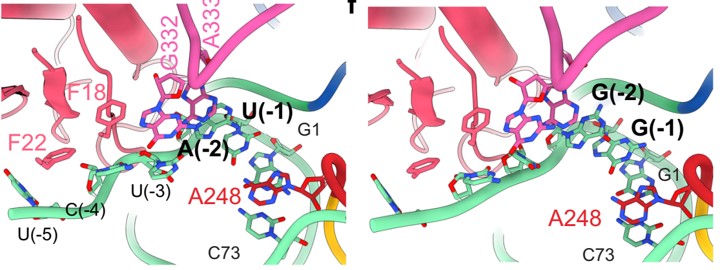
A defining challenge in modern RNA biology is developing quantitative models of RNase specificity that integrate deep mutational scanning, structure, and transcriptomics, that are useful for predicting targets and processing rates in vivo. In collaboration with Dr. Eckhard Jankowsky we developed a robust and adaptable strategy for mapping landscapes of RNase and RNA binding protein molecular recognition that employs deep mutational scanning to obtain foundational kinetic measurements for constructing comprehensive and quantitative specificity models. Our vision is to use this approach to investigate the specificity of new biomedically important RNases, discover how the cellular milieu influences RNA specificity.
Our approach to modeling RNA binding specificity was largely developed using bacterial RNase P as an experimental system. RNase P is an essential tRNA processing endonuclease that removes the 5’ leader sequences of precursor tRNAs (ptRNAs). Our studies showed how local sequence context can direct RNA processing along specific pathways and illustrate a complex specificity landscape even for a comparatively simple, well studied RNA processing endonuclease. In collaboration with Dr. Derek Taylor we used cryoEM to determine the structures of E. coli RNase P bound to alternative ptRNAs which provided a wealth of new insights into its broad specificity. Our long-term studies of bacterial RNase P from ESKAPE pathogens promises to be similarly transformative for understanding RNA specificity as well as advancing drug discovery.
Kinetics and Specificity of SARS-CoV-2 Nsp15 Endonuclease
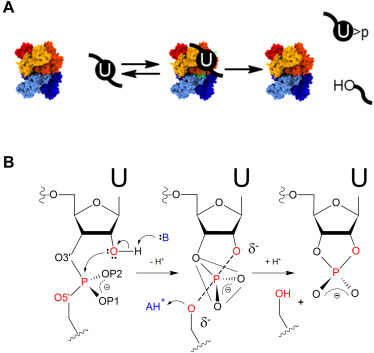
The adaptability of our approach to RNA specificity facilitates application to emerging biomedically important systems. SARS-CoV-2 Nsp15 is a hexameric endonuclease with broad specificity whose range of functions and targets are unknown. Nsp15 is variously proposed to cleave at specific sites in the viral genome to regulate expression, target RNA hybrids to evade innate immunity, and participate in clearance of host RNAs. Recent structural and biochemical studies of Nsp15 show cleavage of both dsRNA and ssRNA substrates, but the functional importance of these observations is limited by lack of quantitative kinetic information.
We analyzed the single- and multiple-turnover kinetics of Nsp15 for model RNA substrates. Biphasic kinetics of ssRNA substrates demonstrates that Mn2+ stabilizes alternative enzyme states that have faster substrate cleavage on the enzyme. The pH-rate profiles in the presence and absence of Mn2+ reveal active-site ionizable groups with similar pKas of ca. 5. An Rp stereoisomer phosphorothioate modification at the scissile phosphate had minimal effect on catalysis supporting a mechanism involving an anionic transition state. These data demonstrated that Nsp15 employs a conventional acid–base catalytic mechanism passing through an anionic transition state, and that divalent ion activation is substrate dependent. With a kinetic framework in hand we are applying the tools of alternative enzyme kinetics to define principles of Nsp15 specificity focusing on the role of RNA structure and dynamics.
The public health importance of understanding SARS-CoV-2 biology cannot be understated, the emergence of new variants, ‘long-COVID’, and ectopic infection raise many unanswered questions about coronavirus biology. Addressing these questions is critically limited by lack of basic functional knowledge of viral proteins including Nsp15. Thus, developing a comprehensive model of Nsp15 specificity using in vitro kinetics, deep mutagenesis, and quantitative specificity modeling will allow us to assess currently proposed functional roles, evaluate rates of cleavage of alternative substrates, and identify potential new in vivo targets.